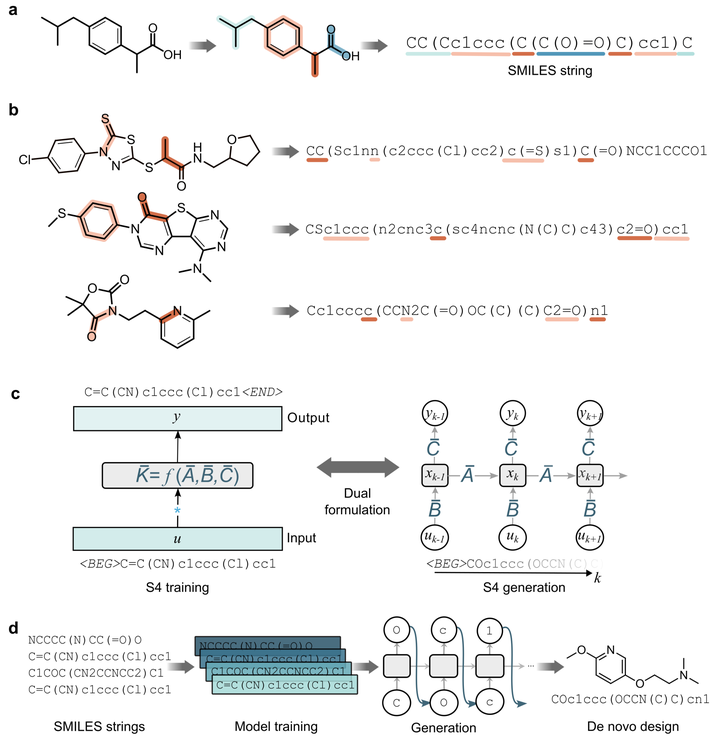
Abstract
Generative deep learning is reshaping drug design. Chemical language models (CLMs) – which generate molecules in the form of molecular strings – bear particular promise for this endeavor. Here, we introduce a recent deep learning architecture, termed Structured State Space Sequence (S4) model, into de novo drug design. In addition to its unprecedented performance in various fields, S4 has shown remarkable capabilities to learn the global properties of sequences. This aspect is intriguing in chemical language modeling, where complex molecular properties like bioactivity can ‘emerge’ from separated portions in the molecular string. This observation gives rise to the following question: Can S4 advance chemical language modeling for de novo design? To provide an answer, we systematically benchmark S4 with state-of-the-art CLMs on an array of drug discovery tasks, such as the identification of bioactive compounds, and the design of drug-like molecules and natural products. S4 shows a superior capacity to learn complex molecular properties, while at the same time exploring diverse scaffolds. Finally, when applied prospectively to kinase inhibition, S4 designs eight of out ten molecules that are predicted as highly active by molecular dynamics simulations. Taken together, these findings advocate for the introduction of S4 into chemical language modeling – uncovering its untapped potential in the molecular sciences.