Exploring Data‐Driven Chemical SMILES Tokenization Approaches to Identify Key Protein‐Ligand Binding Moieties
Abstract
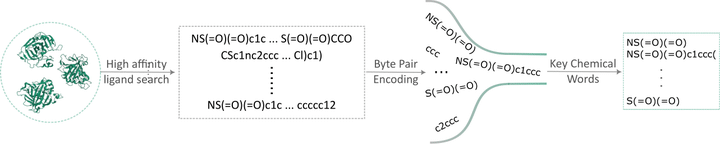
Machine learning models have found numerous successful applications in computational drug discovery. A large body of these models represents molecules as sequences since molecular sequences are easily available, simple, and informative. The sequence-based models often segment molecular sequences into pieces called chemical words, analogous to the words that make up sentences in human languages, and then apply advanced natural language processing techniques for tasks such as de novo drug design, property prediction, and binding affinity prediction. However, the chemical characteristics and significance of these building blocks, chemical words, remain unexplored. To address this gap, we employ data-driven SMILES tokenization techniques such as Byte Pair Encoding, WordPiece, and Unigram to identify chemical words and compare the resulting vocabularies. To understand the chemical significance of these words, we build a language-inspired pipeline that treats high affinity ligands of protein targets as documents and selects key chemical words making up those ligands based on tf-idf weighting. The experiments on multiple protein-ligand affinity datasets show that despite differences in words, lengths, and validity among the vocabularies generated by different subword tokenization algorithms, the identified key chemical words exhibit similarity. Further, we conduct case studies on a number of target to analyze the impact of key chemical words on binding. We find that these key chemical words are specific to protein targets and correspond to known pharmacophores and functional groups. Our approach elucidates chemical properties of the words identified by machine learning models and can be used in drug discovery studies to determine significant chemical moieties.