The surprising ineffectiveness of molecular dynamics coordinates for predicting bioactivity with machine learning
Abstract
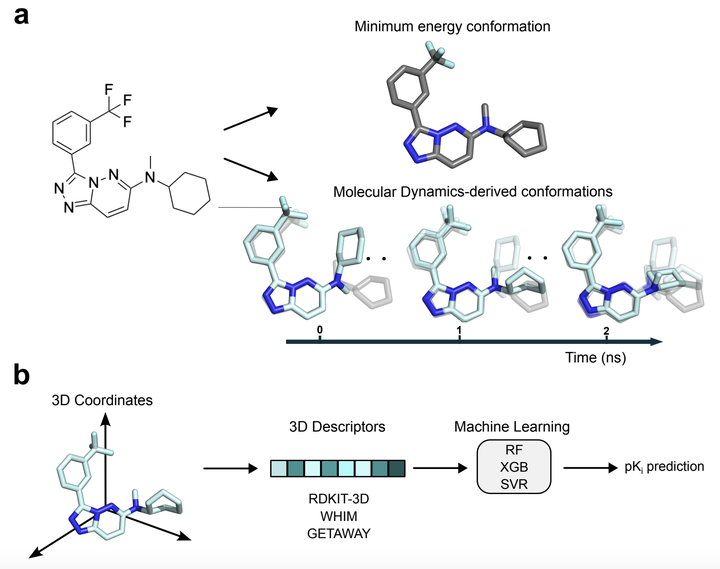
Accurate prediction of protein-ligand binding affinity remains a major challenge in drug discovery, despite the rapid progress of machine learning. Interestingly, machine learning approaches based on two-dimensional molecular information (e.g., binary fingerprints) often outperform those using three-dimensional (3D) information, possibly due to the usage of minimum-energy conformations. This raises questions about how to incorporate more sophisticated three-dimensional information (e.g., ligand flexibility and binding-induced conformational changes) for bioactivity prediction. To this end, we systematically investigate whether coordinates derived from molecular dynamics (MD) can improve prediction performance over minimum-energy conformations. MD-derived coordinates capture dynamic molecular interactions, which are hypothesized to reflect a more realistic representation of ligand-protein binding events. Using over 2600 protein-ligand complexes across three macromolecular targets, we compared multiple machine learning approaches using well-established 3D descriptor sets. Surprisingly, our results show that MD-derived coordinates do not consistently outperform ‘static’ 3D structures, despite their ability to capture dynamic molecular interactions. These findings highlight the persistent challenge of effectively leveraging three-dimensional and dynamic information for bioactivity prediction and underscore the need for improved representations approaches to bridge this gap.